Investigation of the radiation characteristics of the torch. Combinations of temperature and thermal conditions
The partial pressure of each gas that is part of the mixture is the pressure that would be created by the same mass of this gas if it occupied the entire volume of the mixture at the same temperature.
In nature and technology, we very often deal not only with one pure gas, but with a mixture of several gases. For example, air is a mixture of nitrogen, oxygen, argon, carbon dioxide and other gases. What does the pressure of a mixture of gases depend on?
In 1801, John Dalton established that the pressure of a mixture of several gases is equal to the sum of the partial pressures of all the gases that make up the mixture.
This law is called the law of partial pressures of gases
Dalton's Law The partial pressure of each gas in a mixture is the pressure that would be created by the same mass of that gas if it occupied the entire volume of the mixture at the same temperature.
Dalton's law states that the pressure of a mixture of (ideal) gases is the sum of the partial pressures of the components of the mixture (the partial pressure of a component is the pressure that a component would exert if it alone occupied the entire space occupied by the mixture). This law indicates that each component is not affected by the presence of other components and the properties of the component in the mixture do not change.
Two laws of Dalton
Law 1 The pressure of a mixture of gases is equal to the sum of their partial pressures. It follows from this that the partial pressure of a component of a gas mixture is equal to the product of the pressure of the mixture and the molar fraction of this component.
Law 2 The solubility of a component of a gas mixture in a given liquid at a constant temperature is proportional to the partial pressure of this component and does not depend on the pressure of the mixture and the nature of other components.
The laws are formulated by J. Dalton resp. in 1801 and 1803.
Dalton's law equation
As already noted, the individual components of the gas mixture are considered independent. Therefore, each component creates pressure:
\[ p = p_i k T \quad \left(1\right), \]
and the total pressure is equal to the sum of the pressures of the components:
\[ p = p_(01) k T + p_(02) k T + \cdots + p_(i) k T = p_(01) + p_(02) + \cdots + p_(i) \quad \left( 2\right),\]
where \(p_i\) is the partial pressure i of the gas component. This equation is Dalton's law.
At high concentrations, high pressures, Dalton's law is not fulfilled exactly. Since the interaction between the components of the mixture is manifested. Components are no longer independent. Dalton explained his law using the atomistic hypothesis.
Let there be i component in the mixture of gases, then the Mendeleev-Claiperon equation will look like:
\[ ((p)_1+p_2+\dots +p_i)V=(\frac(m_1)((\mu )_1)+\frac(m_2)((\mu )_2)+\dots +\frac(m_i )((\mu )_i))RT\ \quad \left(3\right), \]
where \(m_i \) are the masses of the gas mixture components, \((\mu )_i \) are the molar masses of the gas mixture components.
If you enter \(\left\langle \mu \right\rangle \) such that:
\[ \frac(1)(\left\langle \mu \right\rangle )=\frac(1)(m)\left[\frac(m_1)((\mu )_1)+\frac(m_2)( (\mu )_2)+\dots +\frac(m_i)((\mu )_i)\right] \quad \left(4\right), \]
then equation (3) can be written as:
\[ pV=\frac(m)(\left\langle \mu \right\rangle )RT \quad \left(5\right). \]
Dalton's law can be written as:
\[ p=\sum\limits^N_(i=1)(p_i)=\frac(RT)(V)\sum\limits^N_(i=1)((\nu )_i)\ \quad \left (6\right). \]
\[ p_i=x_ip\ \quad \left(7\right), \]
where \(x_i-molar\ concentration\ i-th \) gas in the mixture, while:
\[ x_i=\frac((\nu )_i)(\sum\limits^N_(i=1)(n_i))\ \quad \left(8\right), \]
where \((\nu )_i \) is the number of moles \(i-th\) gas in the mixture.
Javascript is disabled in your browser.ActiveX controls must be enabled in order to make calculations!
This law shows the additivity property of the partial volume.
v cm = Sv i
v i / v cm \u003d n i / N \u003d y i
v i \u003d y i × v c m
Used in calculations of gas fields.
33. Equation of state of ideal gases, supercompressibility factor
COEFFICIENT OF SUPERCOMPRESSION OF NATURAL GASES
Gases - real and ideal.
Ideal gases are when the interaction of molecules with each other is neglected.
P - absolute pressure (Pa), V - volume (m 3), G - mass of a substance (kg), T - temperature (K), R - universal gas constant (kJ / K × kg).
(for an ideal gas).
z is the degree of deviation of a real gas from an ideal one, or the coefficient of compressibility of a real gas.
34. Van der Waals equation and its physical meaning
The property of ideal gases is that: Р×V/(G×R×Т)=1=z.
The new coefficient z introduced by us, which is equal to 1 for ideal gases, and differs from it for real gases, is called supercompressibility factor.
z is the coefficient by which the properties of ideal gases are applied to real ones. It characterizes the degree of deviation of the ideal gas from the real one.
Various attempts have been made to improve the description:
1) Van der Waals equation:
(P + a / v 2) (v-in) \u003d R × T,
where v is the specific volume; c – correction for the volume of molecules; a / v 2 \u003d const - the coupling constant of molecules.
The value a / v 2 expresses the internal pressure, which is, as it were, the resultant force of attraction of all molecules in the volume v.
At pressures up to 100 MPa and temperatures T=150°C, it is necessary to determine the most accurate description of the dependencies. In considering this issue, science went in two directions:
1. introduction of the supercompressibility factor z;
2. adding additional constants to the equation of state.
2) Any experimental dependence can be described using a polynomial, so the way to increase the number of constants was chosen. The most common were equations with five Beatty-Bridgeman constants and eight Benedict-Webb-Rubin constants. All constants are determined by the least squares method.
35. Reduced and critical parameters of gases and their mixtures
I. Introduction of z into the equation of state. On the basis of experiments, it turned out that if the given parameters P pr, T pr are the same and are in the corresponding states, then such thermodynamic properties as the supercompressibility coefficient are the same for different gases. Those. z=f(P pr, T pr).
Given parameters ideal components - dimensionless quantities showing how many times the actual parameters of the state of gases are greater than the critical ones. The parameters are understood as: P abs, T, V and z.
T CR =T/T cr; P CR =P/P cr; z pr \u003d z / z cr.
The given parameters are calculated on the basis of critical parameters, from here we will consider the issue of determining critical parameters.
 P kr =Sy i ×P kri ; T cr =Su i ×T cri ; z cr =Sу i ×z cri
P kr =Sy i ×P kri ; T cr =Su i ×T cri ; z cr =Sу i ×z cri
Dependences of the given parameters look as follows: z Т pr
36 Dependence of the coefficient of supercompressibility of natural gas on the reduced pressure and temperature
The reduced parameter is a dimensionless value showing how many times the parameters P,V,r more or less critical.
 real gases
- a mixture of hydrocarbon and non-hydrocarbon components. Molecules of argon, xenon, krypton and methane have a spherical configuration. The molecules of such gases as propane and butane are non-spherical, therefore, to take into account the shape of the molecules, the parameter was introduced - acentric factor
(w). He shows that if the molecule is spherical, then the forces that act on it are spherical, which indicates the symmetry of the forces. If the molecules are not spherical, then there is an asymmetry active forces.
real gases
- a mixture of hydrocarbon and non-hydrocarbon components. Molecules of argon, xenon, krypton and methane have a spherical configuration. The molecules of such gases as propane and butane are non-spherical, therefore, to take into account the shape of the molecules, the parameter was introduced - acentric factor
(w). He shows that if the molecule is spherical, then the forces that act on it are spherical, which indicates the symmetry of the forces. If the molecules are not spherical, then there is an asymmetry active forces.
z=z(P pr, T pr, w)
z cm \u003d z 0 (P pr, T pr) + z 1 (P pr, T pr) × w cm,
where z 0 is the supercompressibility coefficient of a simple gas. For a simple gas, the molecules are spherical and w=0.
z 1 is a correction to the coefficient of supercompressibility of a complex gas, which depends on Р pr, Т pr and w¹0.
w cm is the acentric factor of the entire mixture characterized by certain concentrations:
w cm \u003d Sу i ×w i
From here it can be seen that the acentric factor of the mixture depends on the acentric factor of each component.
i is the molar concentration of the component.
37 Density of natural gas and stable hydrocarbon condensate
For natural gas:
r P, t \u003d r P0, t0 × (P × z 0 × T 0) / (P 0 × z × T)
For stable condensate:
r (C 5+) \u003d 1.003 × M to / (M to +44.29)[kg/cm3]
According to the refractive index, determined experimentally, it is possible to calculate:
1gМ to =1.939+0.0019×t to +1g(2.15 - n D),
where t to - the boiling point of the condensate; n D is the refractive index.
These coefficients are empirical in nature.
But the density of a stable condensate can also be calculated using a different formula, namely:
r to =Sх i ×М i /Sх i ×n i /r i ,
where x i is the mole fraction of the i-th component;
r i is the density of the i-th component;
M i is the molecular weight.
________________________
Gas density. The greater the proportion of high molecular weight components in the gas, the greater the molecular weight of the gas, which is linearly related to the density of the gas:
ρcm = Mcm/22.41
Usually ρ is in the range of 0.73 - 1 kg/m3. the density of the individual components of hydrocarbon gases (and hydrogen sulfide), with the exception of methane, is greater than 1.
To characterize the density of the gas, its ratio to the density of air under the same conditions is also used (air density under normal conditions is 1.293 kg/m3).
where is the relative density of the gas; ρcm, ρv are the density of gas and air, respectively. The relationship between the density of a gas and its molecular weight, pressure and temperature is determined by the law of the state of gases, which can be represented as:
38 Viscosity of gas and gas mixtures
Viscosity of gases. The viscosity of a gas depends on its composition, pressure and temperature. The viscosity of gases is due to the exchange of momentum between layers of gas moving at different velocities relative to each other. This exchange occurs due to the transition of molecules from one layer to another during their chaotic movement. Since large molecules have a shorter free path (the probability of their collision with each other is relatively high), the amount of motion they transfer from layer to layer is less than that of small molecules. Therefore, the viscosity of gases with an increase in their molecular weight, as a rule, decreases.
 With an increase in temperature, the speed of movement of molecules increases and, accordingly, the amount of motion transferred by them from layer to layer, therefore, at low pressures, the viscosity of the gas increases with increasing temperature. At high pressures, when the distances between molecules are small, the transfer of momentum from layer to layer changes somewhat. It occurs mainly, as in liquids, due to the temporary association of molecules at the boundary of layers moving at different speeds. The probability of such a combination decreases with increasing temperature. Therefore, at high pressures, the viscosity of gases decreases with increasing temperature.
With an increase in temperature, the speed of movement of molecules increases and, accordingly, the amount of motion transferred by them from layer to layer, therefore, at low pressures, the viscosity of the gas increases with increasing temperature. At high pressures, when the distances between molecules are small, the transfer of momentum from layer to layer changes somewhat. It occurs mainly, as in liquids, due to the temporary association of molecules at the boundary of layers moving at different speeds. The probability of such a combination decreases with increasing temperature. Therefore, at high pressures, the viscosity of gases decreases with increasing temperature.
With increasing pressure, the viscosity of gases increases: slightly at low pressures and more intensively at high pressures.
The viscosity of a gas is determined experimentally by measuring its flow rate in capillaries, the rate of fall of the ball in the gas, the attenuation of the rotational oscillations of the disk, and other methods. The change in viscosity at various pressures and temperatures can be determined by calculation and from graphs depending on the given pressure and temperature.
Gas viscosity at low pressures and temperatures close ideal gas viscosity. So, you can use the kinematic theory by writing the equation for a rarefied gas:
m=r×v×l/3,
where v is the average speed of the molecules; l is the length of the free path.
According to the kinetic theory, viscosity depends on pressure and temperature:
With increasing pressure, the density increases, but l decreases, resulting in an increase in the probability of collision, the average speed of movement is constant, and the viscosity in the initial period is almost constant (Dр<<).
With increasing temperature, the viscosity increases, because the average velocity of the molecules increases, and the density and mean free path practically do not change.
At the same time, from the definition of viscosity, the forces that prevent the movement of one layer relative to another must change, which means that the change in viscosity is complex.
 m P max
m P max
At low pressures, m depends little on the pressure drop. With increasing pressure and increasing temperature, the viscosity of gases (m) decreases.
If we have an increase in the molecular weight of the gas, then the viscosity will increase accordingly.
Accounting for the presence of non-hydrocarbon gases, their effect on viscosity is carried out as follows:
m \u003d y a × m a + (1 - y a) × m y,
where y is the molar fraction;
m a is the viscosity of the non-hydrocarbon gas;
m y is the viscosity of the hydrocarbon gas.
The dependence of m on molecular weight can be graphically depicted:

39. Dependence of gas viscosity on composition and thermobaric conditions
40 Isobaric molar heat capacity of natural gases
Let's consider two main thermodynamic processes: at constant pressure (isobaric) and at constant volume (isochoric).
To calculate the ongoing processes in gases, the concepts isobaric and isochoric specific heat capacity .
C p \u003d (dQ / dT) p
C v \u003d (dQ / dT) v
dQ=di - v×dр,
where i is the enthalpy of an ideal gas.
di \u003d dQ + v × dp \u003d C p × dT + (v - T × (dp / dT) p) dv
With p=const: dQ=di=С р ×dТ Þ С р =(di/dТ) р
That. Cp depends on the temperature.
С ri =0.523×(8.36+0.008×t)m i 3/4[kJ/(kmol×K)]
The heat capacity of real gases is determined by the additivity rule, i.e.:
C pcm \u003d Sу i × С рi
Isobaric molar heat capacity depends on pressure and temperature:
C p \u003d C ri (t) + DC p (p, t),
where DСр is the isothermal correction of heat capacity for pressure and temperature.
 T pr
T pr
The states of hydrocarbon systems are of particular relevance, because are in the region of critical states, where phase transformations take place.
All equations obtained on the basis of the experiment are semi-empirical.
41 Dependence of the isobaric molar heat capacity of real gases on pressure and temperature
42 Pent-Robinson Equation of State
43 The Pent-Robinson equation for the supercompressibility factor
44 Using the Pent-Robinson equation to describe the deviation of the thermo-physical properties of gases
The solution of problems related to the production, transport and processing of gas is associated with the Peng-Robinson equation (1975):
P \u003d R × T / (v-c) \u003d a (T) / (v × (v + c) + c × (v-c)),
where а(Т), в are coefficients determined by critical parameters, and а(Т) is some function.
v is the molecular volume.
z 3 - (1 - B) × z 2 + (A - 3 × B 2 - 2 × B) × z - (A × B - B 2 - B 3) \u003d 0,
where A \u003d a (T) × P / (R 2 × T 2),
V=v×P/(R×T)
If the mixture is in a two-phase state, then the larger root corresponds to the vapor phase, and the smaller root corresponds to the liquid phase.
Under critical conditions, z cr = const is a constant value - and z cr = 0.307. Then:
a(T cr)=0.45724×R 2 ×T cr 2 /R cr
v(T cr)=0.0778×R×T cr / R cr
If the temperature is different from the critical one, then these coefficients depend on T cr:
a(T)=a(T cr)×a(T cr,w);
in (T) \u003d in (T cr),
where w is a dimensionless function.
At T=T cr a=1.
The relationship between a and temperature (T) can be written as follows:
a 0.5 \u003d 1 + m × (1 - T 0.5), m \u003d f (w).
For a mixture, the Peng-Robinson equation looks like this:
a cm (T) \u003d Sу i ×а i;
in cm (T) \u003d Sу i ×in i,
where ai and wi are calculated by the formulas:
and i =0.457×(R 2 ×T cr i 2 /R cr i)×a i ;
in i \u003d 0.0778 × R × T cr i / R cr i
45 Saturated vapor pressure of hydrocarbon systems and their mixtures
46 Henry's law
Solubility of gases in liquids. At high pressures, the solubility of gases in liquids, including oil, obeys Henry's law. According to this law, the amount of gas Vr that dissolves at a given temperature in a liquid volume Vl is directly proportional to the gas pressure p above the liquid surface:
Vg = α∙р∙V (2.8)
where [a] \u003d [m 2 / N] - Henry coefficient , which takes into account the amount of gas that dissolves in a unit volume of liquid when the pressure is increased by one unit.
a \u003d V g / (V f × p)
The solubility coefficient shows how much gas is dissolved in a unit volume of oil with an increase in pressure per unit. The solubility coefficient of gas in oil is a variable value. Depending on the composition of oil and gas, temperature and other factors, it varies from 0.4∙10-5 to 5∙10-5 1/Pa.
To the greatest extent, the composition of the gas itself affects the solubility of gas in oil. Light gases (nitrogen, methane) are less soluble in oils than gases with a relatively higher molecular weight (ethane, propane, carbon dioxide). In oils containing a greater amount of light hydrocarbons, the solubility of gases is higher compared to heavy oils. As the temperature rises, the solubility of gases in oil decreases.
It follows from Henry's law that the greater the solubility coefficient, the lower the pressure in a given volume of oil dissolves the same volume of gas. Therefore, oils with a high content of methane at high reservoir temperatures usually have high saturation pressures, while heavy oils with a low content of methane at low reservoir temperatures have low saturation pressures. The difference in the physical properties of oil in reservoir conditions and on the surface is associated with the amount of dissolved gas.
47 Solubility of gases in oil and water
The solubility characteristics of gas in oil are as follows:
 cm 3 / cm 3
cm 3 / cm 3
Pressure values are plotted along the abscissa, and the amount of gas dissolved in oil is plotted along the ordinate.
The solubility of gases increases with increasing molecular weight of the gas. Consequently, different gas components have different solubility, which means that natural gas will dissolve in natural oil in a complex way.
Solubility depends on the composition and properties of the oil. Moreover, the solubility of gases increases with an increase in the content of paraffinic hydrocarbons, and with a high content of aromatic hydrocarbons.
Slightly soluble gases obey Henry's law better than highly soluble gases.
The nature of the gas affects the solubility of gases in oil to a greater extent than the composition of oil, although in a compressed gas at high pressures a reversible dissolution of oil components occurs, which can be seen in the flattening of the solubility curves of highly soluble gases.
Solubility factor oil gases varies widely and reaches (4-5)×10 -5 m 3 /(m 3 ×Pa).
Hydrocarbon gases are less soluble in oil with increasing temperature.
In addition to the dissolution process, there is a process of gas extraction from oil. Dissolution is associated with geological conditions, and the process itself took place for a long period. And the process of selection is connected with our activity, and it is already short-term.
contact differential
48 Solubility isotherms of natural gases in oils
The amount of gas released depends on the choice of technology:
Gas has been released and is in contact with oil (gas caps);
Gas was released and we removed it from the oil-gas system (with a branch).
The first of these methods of degassing is called contact , or single stage. Second - differential , or stepped (multiple).
If the process is differential, then the amount of gas remaining in the dissolved state in the oil is greater than with the contact (single-stage). This is due to the transition to the vapor phase of methane.
The amount of gas released from oil is characterized by degassing curves. They are obtained experimentally, and each field has its own curve.
 cm 3 / cm 3
cm 3 / cm 3
Degassing factor It is customary to call the amount of gas released from a unit volume of oil with a decrease in pressure per unit.
In some range of pressure degassing does not occur.
If the gas is degassed, then the phase permeability of oil decreases.
49 Contact and differential degassing of oil
 2 types of degassing curves:
2 types of degassing curves:
1) contact type - all the released gas remains.
2) differential type - gas is removed. Characteristic for laboratory conditions.
For diff. Degassing - the amount of gas is greater than with contact.
Degassing curve:
50 Oil degassing factor
The degassing coefficient is usually called the amount of gas released when the pressure is reduced by one.
In addition to oil, there may be a large amount of water in the formation.
51 Solubility of hydrocarbon gases in water
52
53 How thermobaric conditions affect saturation pressure
Oil saturation pressure - the maximum pressure at which gas begins to be released from oil in an isothermal process, under conditions of thermodynamic equilibrium.
 Among other things, the saturation pressure depends on the temperature and increases with its growth.
Among other things, the saturation pressure depends on the temperature and increases with its growth.
If the saturation pressure is approximately equal to the reservoir pressure, and we will inject cold water, then the reservoir temperature will decrease, which means that gas can be released due to pressure reduction.
Stepanova found that with a very slight gas release (hundredths of a percent), a lubrication effect occurs and the phase permeability for oil increases abnormally.
When we irradiate the rock with ultrasound, gas bubbles begin to be released, control over this process will allow us to control the phase permeability. The number of released bubbles depends on the skeleton of the component of the rock, the composition of the reservoir. From this we can conclude that the saturation pressure varies across the reservoir.
54 Compressibility of oil and its characterizing components
Oil has elasticity, which is measured compressibility factor (or bulk elasticity ).
b n \u003d -1 / V × (dV / dp)
It is about (0.4¼0.7) GPa -1 (for oils that do not contain dissolved gas). Light oils containing a significant amount of dissolved gas have an increased compressibility coefficient (b n reaches 14 GPa -1).
b n depends on temperature and pressure, and the higher the temperature, the greater the compressibility coefficient.
 |
When oil from a reservoir rises to the surface, its composition changes, its volume changes.
The volume factor is calculated by the formula:
v \u003d V pl / V money,
where V pl is the volume of oil in reservoir conditions;
V deg is the volume of degassed oil (on the surface).
 The dependence of the volumetric coefficient on pressure is as follows.
The dependence of the volumetric coefficient on pressure is as follows.
To carry out thermodynamic calculations of systems with gas mixtures or solutions, it is necessary to know their composition. The composition of the mixture can be set:
Mass fractions , where 
 - molar mass i-th component, kg/mol; M i– relative molecular weight i-th component; n i- number i-th substance, mol;
- molar mass i-th component, kg/mol; M i– relative molecular weight i-th component; n i- number i-th substance, mol;
for each phase  ;
;
Mole fractions  , where
, where  - the amount of substance of the mixture, mol; for each phase, the sum of the mole fractions of the components of the mixture
- the amount of substance of the mixture, mol; for each phase, the sum of the mole fractions of the components of the mixture  ;
;
Volume fractions, which are equal to mole fractions 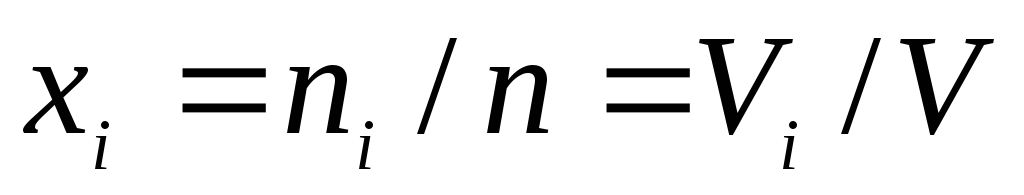 , where
, where  - volume i-th component of the mixture, which at the temperature and pressure of the mixture of gases is called the reduced volume;
- volume i-th component of the mixture, which at the temperature and pressure of the mixture of gases is called the reduced volume;  , m 3 / mol - molar volume of the i-th component of the mixture. In accordance with Avagadro's law, the molar volumes of all components of a mixture of gases are equal and
, m 3 / mol - molar volume of the i-th component of the mixture. In accordance with Avagadro's law, the molar volumes of all components of a mixture of gases are equal and  , where
, where  . The sum of the reduced volumes of the components of a mixture of gases is equal to the volume of the mixture (Amag's law), i.e.
. The sum of the reduced volumes of the components of a mixture of gases is equal to the volume of the mixture (Amag's law), i.e. 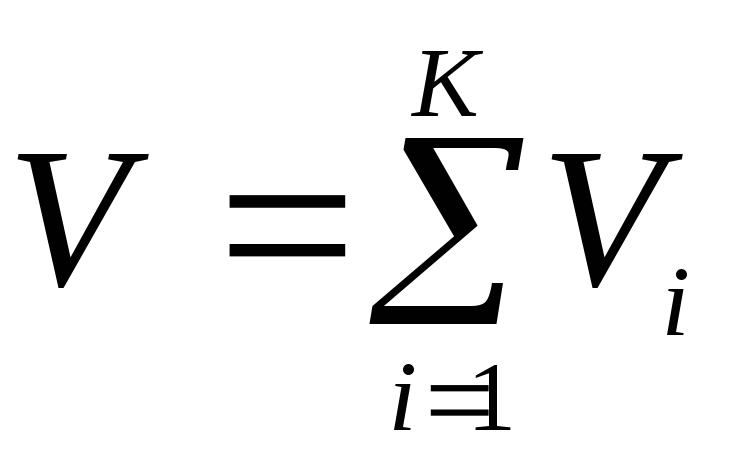 .
.
The composition of a mixture of ideal gases can also be given by partial pressures R i, mass concentrations  and molar concentrations
and molar concentrations  .
.
When setting the composition of solutions, mass and molar concentrations are used.
Partial pressure R i is the pressure i-th component of the gas mixture, provided that it occupies the entire volume intended for the mixture at the temperature of the mixture.
3.2. Relations for mixtures of ideal gases. Dalton's law
The average molar mass of a mixture of gases is given by 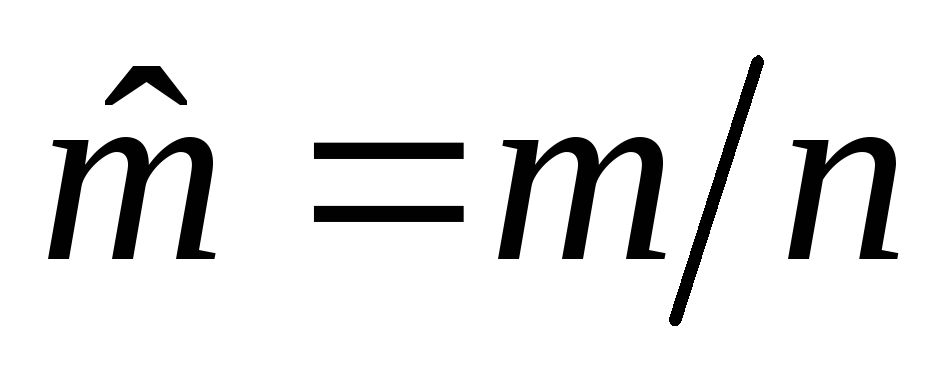 , kg/mol, where
, kg/mol, where  - mass of the mixture;
- mass of the mixture;  - the amount of substance in the mixture. Then
- the amount of substance in the mixture. Then
 .
.
Specific gas constant of a mixture of gases
 , J/(kgK),
, J/(kgK),
where  J/(molK) – molar gas constant;
J/(molK) – molar gas constant;  is the molar mass of the mixture.
is the molar mass of the mixture.
Dalton's law:
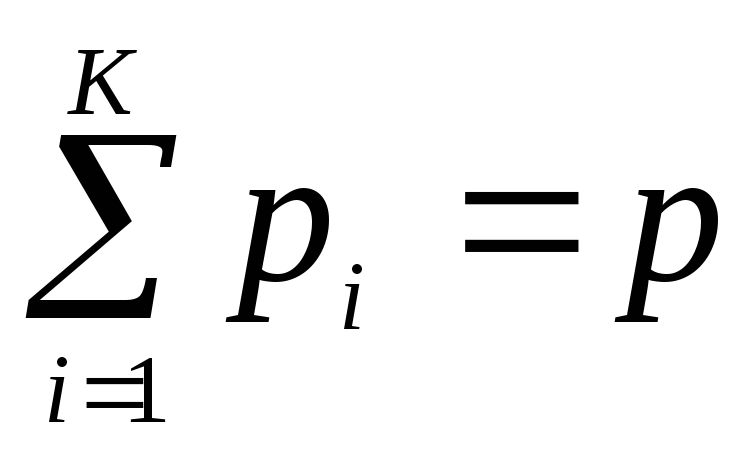 , Pa,
, Pa,
those. the sum of the partial pressures of the individual gases in the mixture is equal to the total pressure of the mixture. Thus, each gas in the vessel occupies the entire volume at the temperature of the mixture, being under its own partial pressure.
The equation of state for a mixture of ideal gases has the form:
 .
.
For partial pressure and for reduced volume i- th component of the mixture, the equations of state have the form:

Then, dividing these equations term by term the first by the second, we have
 .
.
Dividing the equation  to the equation
to the equation  term by term, we get:
term by term, we get:
 .
.
Chapter 4
4.1. Types of heat capacity
Heat capacity is the property of bodies to absorb and release heat when the temperature changes by one degree in various thermodynamic processes. Distinguish between the total average and total true heat capacity.
The total average heat capacity of the thermodynamic process (TP) is the heat capacity of a body with mass m, kg for the final segment of the TP:
 ,[J/K].
,[J/K].
The total true heat capacity of the TP is the heat capacity of a body with a mass m, kg at each given moment TP:
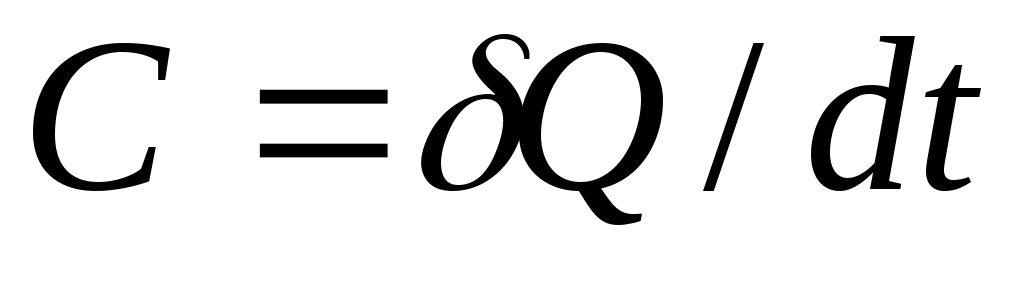 , [J/K].
, [J/K].
Consider an arbitrary TP 1-2 in coordinates  , where Q is the supplied heat in [J]; t
is the temperature in [ 0 C]. Then
, where Q is the supplied heat in [J]; t
is the temperature in [ 0 C]. Then 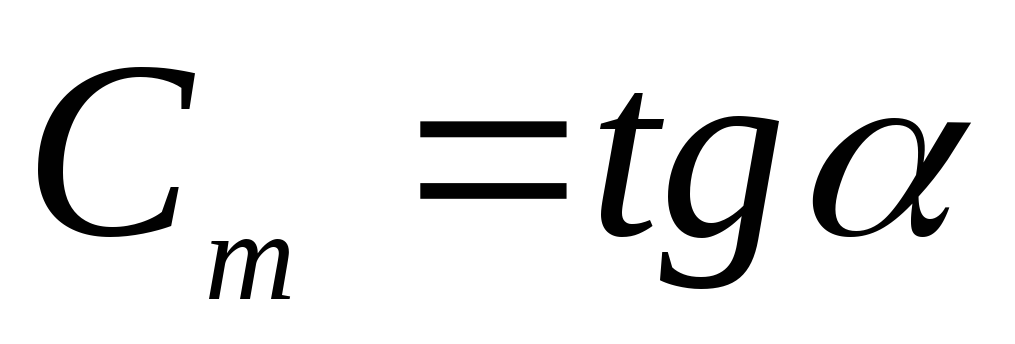 ,
, .
.

If the TS is a homogeneous working fluid, then the relative heat capacities are used in the calculations:
Specific heat capacity - heat capacity per 1 kg of a substance c=c/m, J/kgK;
Molar heat capacity - heat capacity related to 1 mole of a substance  , J/molK;
, J/molK;
Volumetric heat capacity - heat capacity related to 1m 3 of a substance 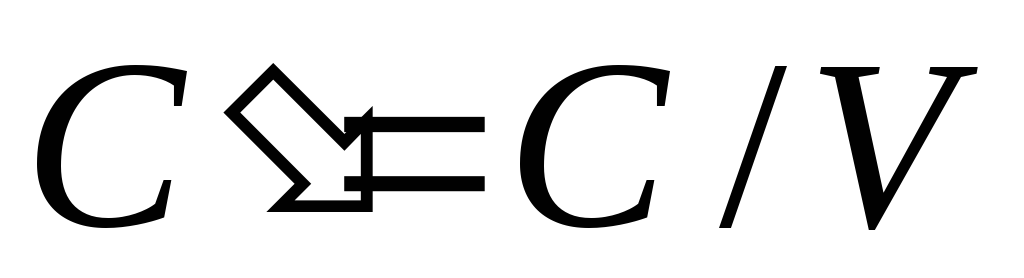 , J / m 3 K.
, J / m 3 K.
Heat capacity is a function of the process and depends on the type of working fluid, the nature of the process and the state parameters. So, the heat capacity in a process with constant pressure is called the isobaric heat capacity:
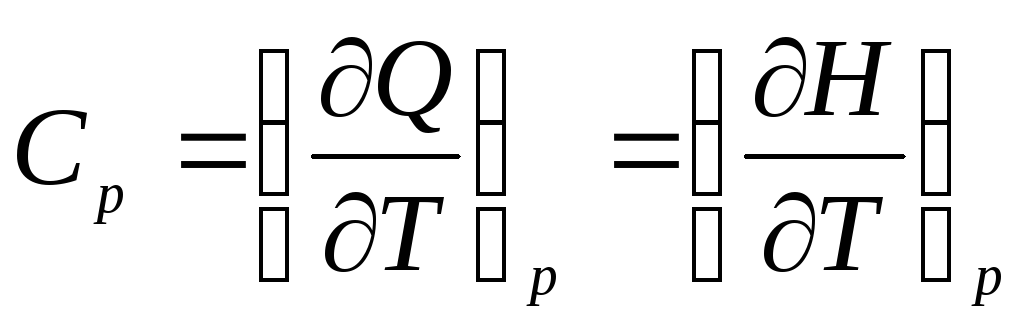 ,
,
where H, J is the enthalpy.
The heat capacity in a constant volume process is called the isochoric heat capacity:
 ,
,
where U, J – internal energy.
The heat capacity of an ideal gas does not depend on temperature and pressure and depends only on the number of degrees of freedom of motion of molecules and, in accordance with the law on the equal distribution of energy over the degrees of freedom of motion of molecules, the heat capacity: 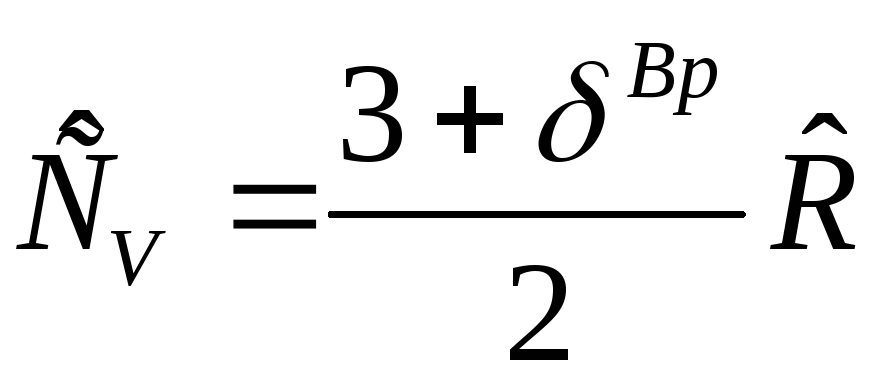 , where
, where  - rotational degrees of freedom, equal to zero for a monatomic gas
- rotational degrees of freedom, equal to zero for a monatomic gas  , for a diatomic gas -
, for a diatomic gas -  =2 and for triatomic gases
=2 and for triatomic gases  =3;
=3; J/molK is the molar gas constant. Heat capacity
J/molK is the molar gas constant. Heat capacity  is determined by the Mayer equation:
is determined by the Mayer equation:
 .
.
For a monatomic gas  and
and  , for a diatomic gas
, for a diatomic gas  and
and  , for three or more atomic gases
, for three or more atomic gases  and
and  .
.
The heat capacity of real gases depends on pressure and temperature. In some cases, we can neglect the influence of pressure on the heat capacity and accept that the heat capacity of real gases depends only on temperature: C= f(t). This dependence is determined experimentally.
The empirical dependence of the specific true heat capacity on temperature can be represented as a polynomial:
where  at a temperature t=0 0 C. For diatomic gases, we can restrict ourselves to two terms:
at a temperature t=0 0 C. For diatomic gases, we can restrict ourselves to two terms: 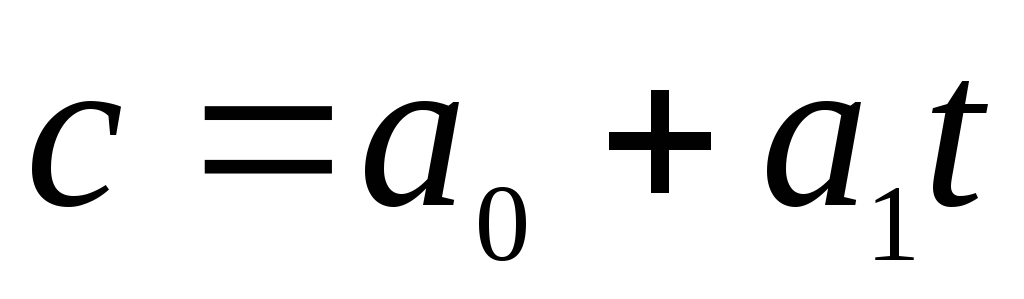 , or
, or  , where
, where 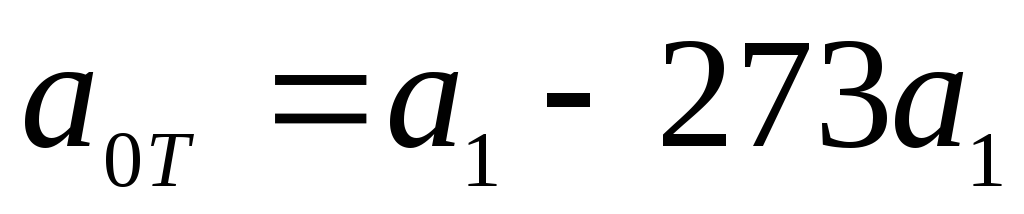 .
.
For the final section of the process 1-2, the amount of heat is:
Then the average heat capacity in this section of the process will be equal to:
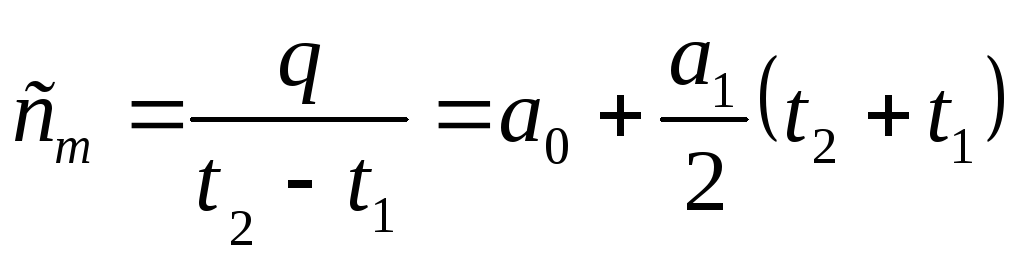 , J/kgK.
, J/kgK.
In the region of low temperatures at T<100К прекращается вращательное движение молекул и колебательное движение атомов, а при температуреT→0K, the translational motion of molecules also stops, i.e. at T=0K WITH R = C v=0 and the thermal motion of molecules stops (experimental data of Nernst et al., 1906-1912). At a temperature T→0K properties of substances cease to depend on temperature, as illustrated in the graph of heat capacity versus absolute temperature.


